

随着基因组学研究的深入,肿瘤液体活检也逐步从实验室过渡到实际临床应用当中。尽管cell-free DNA的概念早在70年前已经被提出,其在癌症诊断中的潜力依然不断地被发掘。近日,在著名期刊Nature Biomedical Engineering上发表的题为“Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics”的综述文章,对肿瘤cfDNA检测领域的现状和未来发表了一些独到见解,本文为大家介绍一下这篇文章的主要内容。

文章发表在Nature Biomedical Engineering上
概 述
血浆中的游离DNA(cfDNA)来源于细胞受损破裂后释放的DNA,经由核酸酶裂解成约160nt大小的片段,其产生和清除是一个动态过程(半衰期约为5~150分钟)。而这种“全局快照”能力使其成为许多疾病的理想分子标志物。作为对影像学和组织活检的补充手段,近年来,cfDNA检测被越来越多的应用到肿瘤诊断领域。肿瘤基因组通常会携带特征性的基因突变位点,而准确获取cfDNA中特定位点突变型与野生型所占的比例,也就是变异等位基因频率(VAF)可以衡量循环肿瘤DNA(ctDNA)的含量水平。然而,cfDNA长度短、含量低,而其中属于ctDNA的部分更是微乎其微,对检测技术的灵敏度以及候选位点的选取都是不小的考验。目前临床上已经可以看到各种相关的检测设备,然而没有哪种单一的技术可以兼顾可靠性、便捷性以及较低的成本。这篇综述讨论了现阶段肿瘤cfDNA检测领域面临的技术挑战和机遇。
综述内容
cfDNA检测在癌症管理中的角色
尽管FDA批准的基因检测产品尚在少数,但越来越多的指南倾向于将这类技术纳入到癌症管理当中。以肺癌为例,cfDNA的检测可以应用到各个诊疗环节(图1)。约有70%的非小细胞肺癌(NSCLC)患者在出现明显症状后被诊断为晚期(III或 IV),只有不到三分之一的患者很幸运地通过主动体检等方式提前发现并在早期干预。目前,cfDNA检测主要的应用场景是帮助占大多数的晚期患者进行治疗决策,clinicaltrials上登记的相关临床试验超过了100个。除了针对某些基因(如EGFR)指导靶向用药之外,通过检测血液肿瘤突变负荷(bTMB)或者微卫星不稳定(MSI)预测免疫治疗效果也是当下的热门方向。随着技术不断改进,cfDNA还可用于治疗(尤其是联合治疗)后的监测,比如检测与复发或耐药相关的突变,目前在多个癌种(乳腺癌、肠癌、肺癌等)均有相关研究正在开展,逐步实现临床落地。
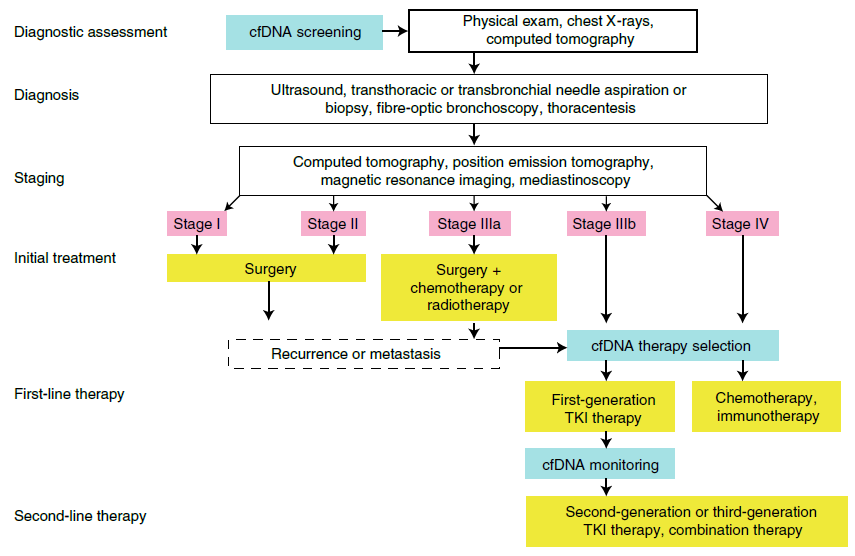
图1. cfDNA检测用于 NSCLC的临床诊断和治疗工作流程。
cfDNA检测也逐渐出现在癌症早筛的研究当中,例如肠癌和鼻咽癌。有研究显示,健康人群的cfDNA中也会存在癌症相关的突变,使得依赖变异检测的方式提高灵敏度和特异性变得十分困难,考虑到筛查的成本及风险因素,效果可能会不尽人意。相反,cfDNA的甲基化检测用于早筛正被寄予厚望[1]。类似TMB之于点突变,基因组甲基化水平是一个整体的肿瘤标志物,并不考虑单个位点的表观标记。事实上,结合单体型等信息还可以辅助判断原发灶的位置[2]。
当前,很多临床试验使用基于低重(low-plex)PCR的突变检测技术将cfDNA作为分层标志物,随着高通量测序技术的成熟,GRAIL、Guardant Health、Foundation Medicine等公司也将针对cfDNA的NGS panel带到了临床试验当中。
分析前的限制因素
好的开头是成功的一半,DNA质量的好坏直接决定结果的准确性,主要涉及样本生物学属性以及前处理过程。生物学因素对cfDNA产量及特性的影响往往不受控,比如,取尿液样本前大量饮水,分析对象受运动和病菌感染而影响的健康状态,肿瘤恶性程度以及对循环系统的可及性等等。相比之下,采样及样本处理过程显得更可控,从而最大程度提高结果的可重复性。理想情况下,静脉采血之后需要马上离心分离血浆和白细胞层,紧接着迅速从血浆中提取cfDNA并进行后续的数字PCR或NGS实验。然而,实际情况往往还要考虑运输、分管、储存等因素,随之而来的风险包括cfDNA的化学损伤以及白细胞污染(图2)。无论体内体外,血液中的DNA都会经历水解、脱氨基以及氧化损伤(已经鉴定的损伤类型超过20种[3]),这些化学反应带来的碱基变异会干扰目标突变位点的检测。虽然有损伤修复产品(比如NEB的preCR)可以选择,但依然难以避免这种假阳性结果的出现。因此,高特异性、高效率逆转损伤的方法对于cfDNA检测将是革命性的。
在采血管中,白细胞缓慢破裂的同时会将自身基因组DNA释放到血浆层。这无疑增加了野生型基因组来源的背景噪声,进而影响VAF的准确检出,尤其是对于低丰度突变。全血可以在室温稳定保存1~7天,随后,血浆层的白细胞污染将会明显增加,一些特殊的保存管可以延长这个期限。因此,提高白细胞稳定性,或者从干扰核酸中区分cfDNA的方法也可以提高最终检测结果的准确性和重复性。
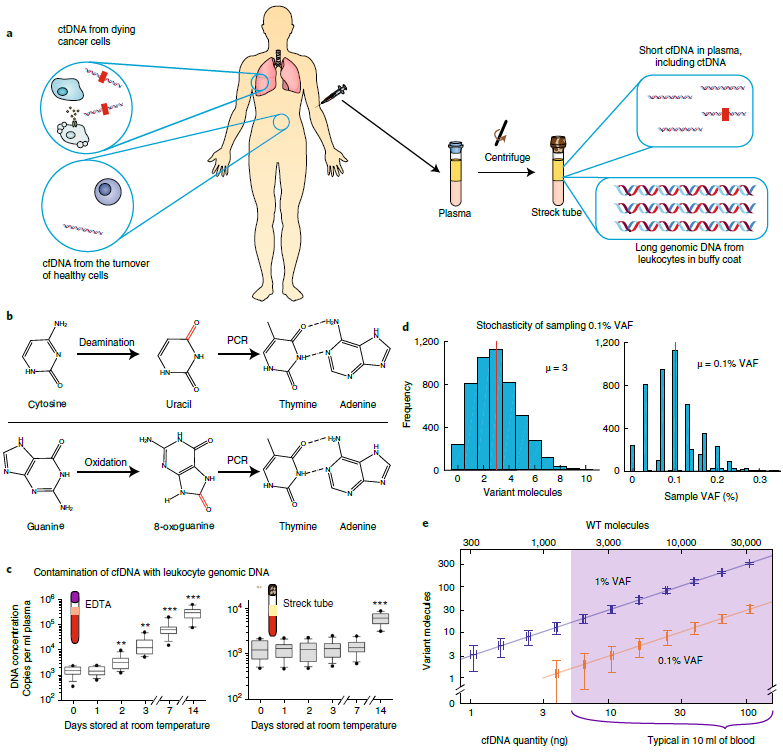
图2.预分析因子影响cfDNA分析的准确性。
微量的cfDNA以及低水平的突变频率意味着泊松抽样统计会得出不准确的VAF值,这与下游使用何种检测技术无关,唯一的解决方法就是提高cfDNA的量。正因为如此,许多商业公司要求最少提供两管10ml的血液样本,以保证在1%及0.1% VAF水平分别达到近乎100%以及超过90%的灵敏度,听起来似乎不难,但实际情形往往难以预料。尿液作为cfDNA来源是一个可行的替代品[4],但从大体积尿液中提取cfDNA也是不小的挑战。并且,尽管基因组污染程度比血浆来源的低,但尿液中的cfDNA片段长度却更短,增加了后续提取以及PCR扩增的难度。总之,可靠且低成本的cfDNA提取方法将是尿液cfDNA应用的突破口之一。
cfDNA的低重分析方法
qPCR、化学发光、等温扩增、转录介导的扩增等技术是典型低重核酸分析方法,他们的共同点是输出单一的治疗决策。肿瘤涉及到众多的生物学通路以及治疗方法,DNA突变状态不仅预示某种疗法是否有效,也能表征治疗反应以及耐药情况。因为单次可获取的cfDNA很少,采用低重分析方法分管检测不太现实,因而这种方式仅限于特定场景(如 EGFR T790M指导三代EGFR TKI的使用)。当前,数字PCR(ddPCR)方法相比qPCR更灵敏(0.05% VS 1% VAF),定量也更准确,但也存在价格高、设备难以普及等缺点。成本相当的NGS技术可以实现更高重数的检测,反而在临床推广上走在了前面。其它低重检测技术包括电化学、CRISPR介导的等温扩增、纳米微粒、单分子荧光等都具有很高的灵敏度。未来,更高的检测通量将是cfDNA检测应用于临床的重要助力。在中等通量的技术中,MassARRAY可以在单个反应中检出超过40种低丰度(0.1% VAF)的突变[5],以及后续成本低,周期短等优势使其接近肿瘤检测中多个靶向位点同时检测的需求。当然,首次设备投入的花费也是一个相对比较大的数字。
cfDNA的NGS分析方法
当下,NGS技术在研究和临床上应用的已经十分普遍,检测通量在众多分析方法中遥遥领先,以Illumina平台为例,单次检测的分子数(reads)可达1010数量级。同时因为短读长以及较好的准确性和较低的后续成本,Illumina检测平台非常适合应用到cfDNA的分析当中(图3)。人类基因组有3G大小,而肿瘤中已知有价值的基因不过千余[6]。如果测序reads随机覆盖所有cfDNA分子,有用的序列信息将只占很小一部分,超过99.9%的reads将被白白浪费。靶向测序可以很好地解决这个问题,主流思路包括探针捕获以及多重PCR。
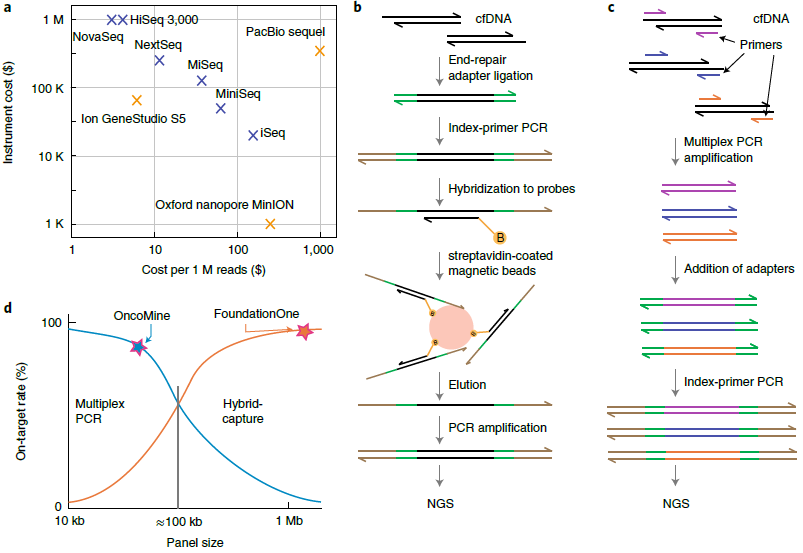
图3. cfDNA的NGS分析。
一个典型的基于连接法的杂交捕获(ligation-and-hybrid-capture)文库构建流程通常分以下几步:cfDNA首先进行末端补齐和加‘A’反应,通过连接酶连上测序接头,经过几轮PCR扩增在末端加上Index序列后使用定制化探针“钓”取目标区域的序列。由于探针杂交可能出现非特异性,这个钓取过程难以做到百分百精准。相对的,基于多重PCR的靶向富集方法借助定制化的引物将目标区域扩增出来,测序接头和Index序列包含在引物中或者同样基于连接法完成[7]。同样的,多对PCR引物同时存在会形成二聚体,扩增过程也会产生非特异产物。所不同的是,二聚体以及部分非特异的扩增子可以通过片段大小筛选去除,并且由于PCR的放大效应导致了更高的on-target rates。因此,针对小型panel,扩增子法会是更好的选择。如果panel覆盖区域超过100kb,杂交捕获法的on-target rates反而更高。这两种建库方式存在以下三个痛点:PCR扩增和NGS过程产生的错误带来假阳性结果;富集过程中的序列损失带来假阴性结果;测序的不均一性可能降低灵敏度或增加成本。PCR引入的突变难以避免并且无法预测,使用高保真PCR酶或者Q ≥ 30的过滤条件也不足以抹平,进而很难判断低于1% VAF突变的真实性。
文库的转化效率(最终可体现的cfDNA占所有cfDNA的比例)与所采用的建库方法关联很大。举例来说,一管10ml的血液可能只含有个位数的目标突变分子,低转化效率意味着很大概率会出现假阴性的结果。Panel设计者倾向于依赖初始cfDNA可测量的最低分子总量来统计特定位点的数目,而测量DNA浓度的方法,比如荧光染料法(Qubit)、吸光度法(Nanodrop)或者ddPCR,根据片段大小、序列、单链核酸等的不同,彼此之间的误差可能超过两倍。因此,目前计算转化效率的方法都存在一定缺陷。具体来说,基于连接法的杂交捕获受限于末端补齐和连接反应的转化效率,一些依赖双端接头提高精度的测序方法(如DuplexSeq[8])受影响可能更大。据报道,当前技术可以达到的转化效率在10%到60%之间[9]。对于多重PCR,瓶颈在于双端引物同时能结合到单条cfDNA分子上的概率。对于160nt长度的cfDNA,100nt的扩增子设计理论上最终的转化效率为37.5%,更长的扩增子意味着这个数值会更低,而对于外泌体来源的ctDNA(超过2,500nt)效果可能更好[10]。此外,以往的DNA提取方法往往会漏掉超短长度的DNA片段,近期研究显示,血液中这种类型的ctDNA也广泛存在[11]。
序列相关的杂交动力学特性影响探针捕获以及PCR的扩增效率,最终导致各位点的测序深度不均一。即使反应条件和引物探针长度相同,杂交动力学速率常数的波动范围也可能会超过三个数量级。调整各引物探针的比例可以一定程度平衡这种偏差,对于商业化NGS panel来说,测序深度的平均值与最低值之间5~50倍的差异都是正常的。与碱基错误及转化率低不同,测序不均一性最主要影响是相同灵敏度下的测序成本。比如,200x的测序深度可以满足最低5% VAF的可靠检出,实际可能需要1000x的平均深度才能以这个灵敏度覆盖所有待测位点。因而,提高测序均一性可以显著降低检测服务的成本。
唯一分子标签技术
癌症患者的cfDNA中,相关突变的VAF范围在0.01%到10%之间。较低的VAF提示疾病可能正处于早期,但也可能是亚克隆(subclonal)突变只存在部分肿瘤细胞中。这种突变往往与耐药相关,即使含量很低也可能伴随用药过程迅速扩展导致治疗失败。临床应用当中,对0.1%甚至更低VAF突变的检出是有意义的。唯一分子标签(UMI)被较多地应用到这种超低灵敏度场景以克服建库和测序过程中碱基错误带来的偏差[12],比如NanoSeq的错误率可低至10-9,SaferSeqS可以从十万个模板分子中检测出一个分子的突变。借助UMI突破测序误差下限的核心理念是,将每个原始cfDNA片段都接上序列唯一的标签,通过标签可以从测序结果中判断来源于同一分子的reads,也即“UMI Family”(图4)。最终的过滤识别以UMI Family为单位,真实突变出现的频率总是比随机突变要高。这种策略可以用于杂交捕获法和扩增子法,但随着重数增加,后者的难度相对更大一些。
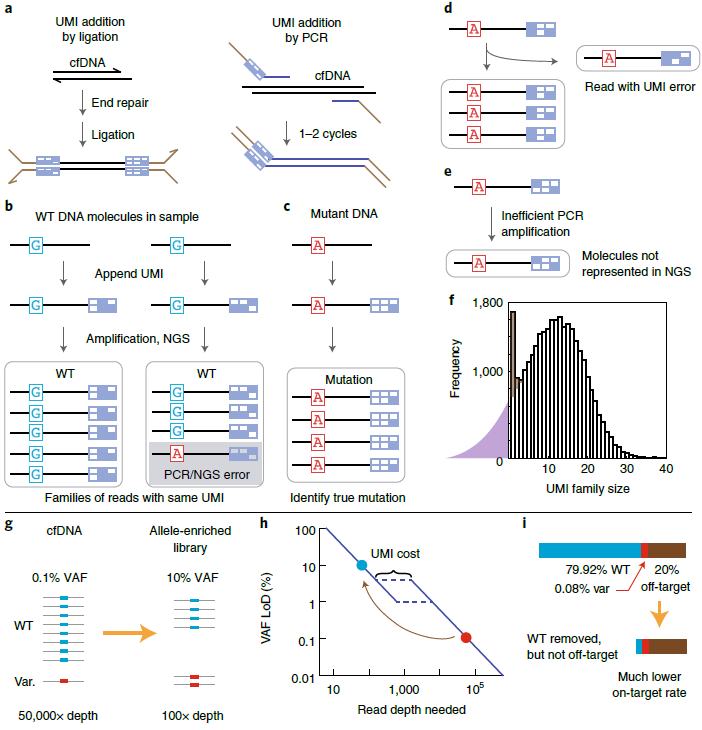
图4.精确检测低于1%的VAF的方法。
显而易见,每个UMI Family的数量越多,误差校正的效果也会越好,带来的问题就是,测序深度的要求也更高(成本最少提高5倍)。除了成本增加,UMI方法也存在一些技术上的限制。首先,cfDNA的起始量要求与常规NGS不同。对于一个平均深度为30,000x的panel测序,如果起始量为10ng,UMI Family的大小等于10,如果是50ng的起始量,这个数值会降低到2。其次,UMI可能会对PCR效率带来难以预测的影响。更糟糕的是,标签序列本身也会存在PCR或测序误差,增加了归类这些reads的难度。常规分析流程会主动忽略reads数低于5(或3)的UMI Family以提高检测和定量的精度,但同时也丢掉了一部分原始cfDNA的信息,导致转化率甚至低于常规NGS方法。一个平均大小为12的UMI Family,其有效转化率会降低接近30%。可以看出,UMI Family越小,原始样本损失的信息也会越多。此外,样本回收比例(成功连接UMI的序列占比)也会影响最终的转化率,基于PCR的连接策略可以获得更高的回收比,例如QAseq(NuProbe Inc.)技术,其半巢式PCR的设计在提高接头连接效率的同时也降低了非特异性扩增的发生。
等位基因富集技术
等位基因富集是一种着眼于从上游提高VAF的文库构建策略,对于放大后的目标突变,常规的低深度测序即可轻松应对。与UMI技术相比,这种策略可以在保持较高灵敏度的同时降低测序成本。实现这一目标的思路有两种:清除野生型序列或者选择性扩增突变序列。例如,振荡电泳(oscillatory electrophoresis)可以通过迁移率区分单碱基的差异,进而有效清除野生型序列[13]。近年来,野生型特异的探针以及核酸酶也被用于实现类似的效果。相对的,选择性扩增突变序列一般是基于在PCR时抑制野生型序列扩增的方法,比如blocker PCR、锁核酸(肽核酸)PCR、ICE-COLD PCR以及抑制探针置换扩增(BDA)法。
这类尚未大规模应用的技术有以下几个痛点。首先,多重性能有待验证。鲜有报道超过20重的等位基因富集技术,然而,当前最小的cfDNA NGS panel也能做到50重(超过1,000重也很常见)。其次,准确定量的前提是对VAF稳定且可重复的富集放大。最后,中靶率低的问题也有待突破。根源在于,随着野生型序列比例降低,脱靶序列(引物二聚体、非特异性扩增产物等)的占比也在上升。在众多技术中,基于吉布斯自由能设计的BDA方法具有更大的可扩增温度区间,因此多重性能表现更优异。高度可控的杂交热动力学过程也使得BDA技术有着不错的灵敏度和稳定性[21]。
cfDNA的非突变标志物
前面章节着重讨论了突变(包括小于50nt的InDels)相关的检测,对于其他几种常见的肿瘤基因标记物,cfDNA的表现如何呢?融合与突变类似,都是“质量性状”的改变,区别在于某一融合型的断点可能出现在不同位置(图5)。比如,NSCLC患者的ROS1基因可能在其总长达140,000 nt的44个内含子的任意一个中发生融合事件。而一个针对cfDNA突变检测的NGS panel可能才100kb,如果要尽可能覆盖融合发生的位置,性价比显然不高。一些NGS panel产品使用热点位置替代全长融合检测,但也牺牲了一部分灵敏度。因此,基于mRNA的融合检测方法更为常见[14]。
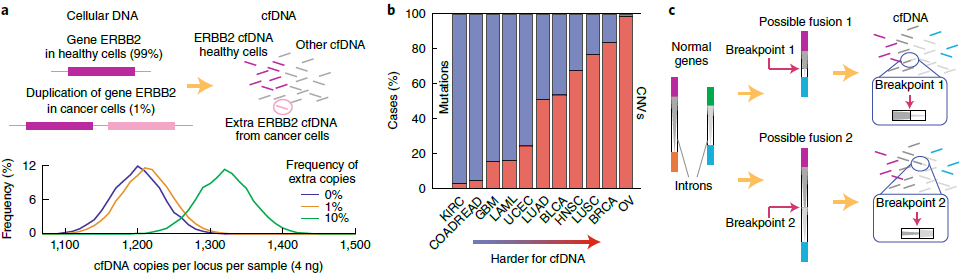
图5. CNV和基因融合是cfDNA分析中比较有挑战性的生物标志物。
CNV的检测需要准确定量出目标基因相对其他基因的增多或者减少,而拷贝区域的末端通常都位于DNA的重复区域,因而无法依靠特殊位点的序列信息进行计算。cfDNA中来源于肿瘤的部分比例可能低于1%,并且受泊松分布影响存在更多不确定性:10ng cfDNA(常规提取可获得的量)约等于3,000个基因组,所以每个位点的DNA分子数量大约为55个,对应的CNV检测下限约为2%。较长的基因更容易检测,因为多个不重叠位置的检测结果可以相互验证。然而,添加UMI的技术难点加剧了泊松统计带来的局限性,当前基于cfDNA的商业化CNV检测产品能做到的极限约为20% VAF[15]。染色体的非整倍性类似于CNV,但由于跨越的区间很大所以检测难度也更低。因此,无创产前诊断(NIPT)可以实现约4% VAF的灵敏度[16]。尽管这种非整倍性在肿瘤中也有发生,但在临床应用中往往不做考虑。
肿瘤早筛对精度的要求
cfDNA用于早筛是当前研究的热门方向[17],GRAIL公司过去3年已经投入超过14亿美元用于相关技术开发和临床试验。接下来我们会探讨该领域在生物学、统计学以及社会层面面临的挑战。
生物学层面,一个关键的问题是,相当比例健康人群的cfDNA中也存在低水平的肿瘤相关突变。这些突变可能来源于克隆造血、体细胞突变或者嵌合。相对的,因为早期肿瘤的cfDNA很难进入到循环系统或跟早期肿瘤相关的致病突变还没有研究清楚,早期癌症人群cfDNA中的肿瘤相关突变也可能无法被检测出来。因此,基本做不到100%的灵敏度和特异性(图6)。对于一个实际癌症发生率为0.5%的筛查人群,假设检测灵敏度和特异性分别为80%和90%,那么阳性预测值(PPV)理论上为3.9%。阳性结果的检测对象一般会被建议接受进一步的检查,大量非必要的二次检查会给受试者带来各方面的负担。因此,基于cfDNA的早筛方法在应用中需要达到接近100%的灵敏度和特异性。通常的做法是,综合受试者的其它条件因素来缩小确认范围,比如年龄、家族史等。作为例子,肠癌早筛产品Cologuard已通过FDA批准,临床灵敏度和特异性分别为94%和87%[18],其推荐的受试者年龄为50岁以上,同时后续的肠镜检查也是相对安全和便宜的。其它相关尝试比如鼻咽癌患者的EB病毒检测,可切除肿瘤的蛋白标志物联合检测[19]等等。全局低水平甲基化以及特定启动子的超甲基化[20]也是目前研究比较热门的多癌症早筛标志物。就不同癌种的早期筛查而言,更低的发病率以及更高的为了确诊而带来的医学风险(尤其对于胶质瘤等脑部肿瘤以及胰腺癌)意味着更低的社会意义和经济价值。
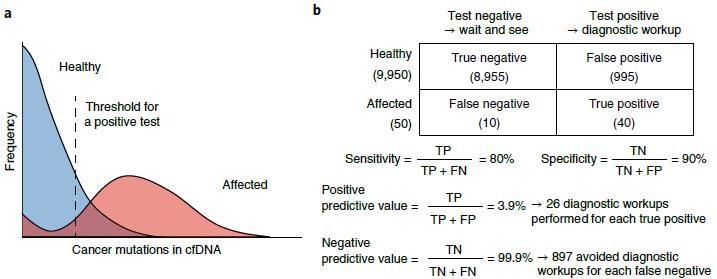
图6.通过cfDNA进行肿瘤早筛的准确性要求。
展 望
从70年前发现至今,cfDNA已经开始影响癌症管理,全球范围众多正在进行的临床试验将其纳入诊断评估环节,但仍然有许多技术层面的机遇和挑战有待研究和开发。相关技术路线大致可以分为快速且低重的检测方法以及成本更高且周期长的高通量测序方法。而大部分cfDNA的诊断应用都需要辅助治疗决策以及检测多重标志物以提高临床灵敏度,基于NGS的手段无疑更胜一筹。如果综合考虑生物学、统计学、医学、物理学、化学、经济学等诸多约束,目前的商业化cfDNA panel只触及了大量潜在信息的一隅,未来仍有广阔的探索空间。相比组织活检以及影像学,非侵入性的肿瘤诊断技术大有可为,前者更契合高危或者病灶已知的人群。除了可以轻松实现多次采样,液体活检在早筛领域也崭露头角,以助力更精准的患者分层。如果局限于突变位点的检测,基于cfDNA的早筛很难突破灵敏度和特异性的瓶颈。融合以及CNV以外,更多来源的标志物(如cfRNA、外泌体、甲基化谱、蛋白水平、CTC等)有待深入探索。多种标志物联合使用或许可以作为突破口。当然,样本处理和分析技术的改进也是其中的关键。


